x 1000 KDa
An origin of replication of C.trachomatis is not apparent using the guideline of proximity to the dnaA gene (there are two dnaA coding sequences in trachomatis). Stephens et al. (xxx) arrived at a probable origin and selected a start for the coordinate system using three measurements that reflect an abrupt change in the chromosomal composition. This is a bit awkward as the starting coordinate falls inside a predicted coding sequence, albeit a hypothetical (CT875). Until this situation is corrected, thi s database adheres to the Stephens proposal and system, remaining consistent with GenBank entries (but differing from the system applied to trachomatis on the PEDANT site).
The average G + C content of the genome is 41.3%. The codon usage as presented on the CUTG Web site (http://www.kazusa.or.jp/codon/) (Nakamura, Y., Gojobori, T. and Ikemura, T. (1997) Nucl. Acids Res. 25, 244-245.) is as follows (based on 50832 codons):
fields: [triplet] [frequency: per thousand] ([number])
UUU 25.3( 1288) UCU 31.9( 1622) UAU 15.2( 771) UGU 11.6( 589)
UUC 16.1( 816) UCC 10.4( 528) UAC 10.5( 536) UGC 7.7( 391)
UUA 29.2( 1484) UCA 7.9( 400) UAA 2.0( 101) UGA 0.5( 27)
UUG 20.2( 1025) UCG 5.5( 282) UAG 0.8( 42) UGG 9.9( 501)
CUU 18.7( 952) CCU 22.1( 1125) CAU 10.6( 540) CGU 13.9( 708)
CUC 7.4( 376) CCC 3.7( 188) CAC 4.9( 247) CGC 6.9( 352)
CUA 9.7( 495) CCA 9.9( 503) CAA 25.8( 1309) CGA 7.2( 364)
CUG 7.5( 381) CCG 2.6( 134) CAG 10.8( 551) CGG 3.4( 175)
AUU 32.2( 1637) ACU 23.4( 1190) AAU 27.2( 1385) AGU 9.0( 455)
AUC 18.4( 935) ACC 7.2( 366) AAC 13.7( 695) AGC 9.6( 490)
AUA 11.5( 584) ACA 23.2( 1178) AAA 52.9( 2690) AGA 15.5( 789)
AUG 21.0( 1069) ACG 7.1( 361) AAG 20.0( 1016) AGG 3.0( 150)
GUU 31.4( 1596) GCU 42.9( 2181) GAU 34.7( 1764) GGU 14.3( 726)
GUC 8.7( 444) GCC 8.6( 436) GAC 14.2( 720) GGC 9.0( 455)
GUA 22.0( 1120) GCA 25.0( 1270) GAA 43.0( 2185) GGA 28.9( 1470)
GUG 12.4( 630) GCG 8.3( 423) GAG 20.2( 1028) GGG 11.6( 591)
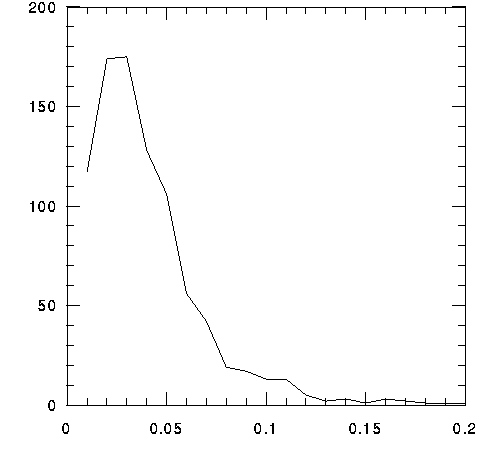
Current analyses of this database show 405 identifiable proteins and 123 conserved but uncharacterized proteins; the remaining ORFs are hypothetical. The original assessment of ORFS by L. Aravind, R. Tatusov, T.Brettin and E.Koonin at the NCBI consider ed frames greater than 240 nucleotides: these were BLASTed against the NR database using gapped BLAST. A subset for which there was high certainty was used with GLIMMER (Interpolated Markov Model) to build a model from which ORFs could be better predicted . ORFs without significant BLAST scores and without significant prediction from GLIMMER were discarded. Taking these ORFs as our starting point, each has been analyzed with respect to 1) similarities outside the genome (orthologs) using gapped BLAST and P si-BLAST, 2) similarities within the genome (isologs and paralogs) using gapped BLAST, 3) similarities to COGs (clusters of orthologous groups) and 4) similarities to databases that emphasize domains, i.e. BLOCKS and ProDom (Documentation). Of the 405 identified proteins, 244 have tentatively been assigned an EC number. The average molecular weight (length) of the 877 proteins inferred from predicted ORFS is 39.6 kDa ( 355 aa); the median molecular weight (length) is 32.8 kDa ( 293 aa). The average and median estimated pI values are 7.44 and 7.12 (hence trachomatis is the most acidic of the three bacteria currently under study).
MOL WT DISTRIBUTION FOR TRACHOMATIS
x 1000 KDa
Of the 877 predicted proteins, 103 appear to be secreted, 241 have transmembrane domains, 156 have coiled-coil regions as determined by suite of structural analysis programs in the SEALS package available from NCBI/NIH. Of 103 proteins predicted to have a signal peptide, 23 had a best hit to E.coli whereas 19 had a best hit to B.subtilis (by criteria stated in the following paragraph). Hence, the majority of secreted proteins were not that similar to either B.subtilis or E.coli. Signal peptide pred iction for C.trachomatis was based solely on both the B.subtilis (gram positive) and E.coli (gram negative) models. Of the 877 proteins, 170 have similarity to a PDB database sequence using BLAST2. A hit requires an E value better than (lower than ) 0.000 1. A higher fraction might be shown to be homologous to PDB sequences using Psi-BLAST and filtering (Huynen, M. et al., J.Mol.Biol. in press).
When C.trachomatis proteins are compared to E.coli and B.subtilis proteins, using BLAST2, 195 are found to have a better hit to E.coli proteins, whereas 259 have a better hit to B.subtilis proteins, consistent with the finding that Chlamydiae are neith er gram positive or gram negative. In this analysis, the E value for the best hit to a genome must be 100 times smaller (better) than the E value for the second genome. Also, only E values of 1e-03 and lower are considered. Note that approximately 197 of the identified and conserved pallidum proteins (651) do not have a best hit to one of these bacteria by these criteria. Best hits for E.coli and B.subtilis fall to 138 and 198 when archaeal and eukarotic sequences are included in the study, representing 4 4 and 88 hits respectively. For a discussion of possible horizontal transmission in C.trachomatis, see Stephens et al. (XXX).
When C.trachomatis proteins are compared to T.pallidum and M.genitalium proteins, 68 have a best hit to M.genitalium and 286 have a best hit to T.pallidum, using the criteria stated above (E value of 1e-03 or better and E value 100 times or more smalle r than the next best hit). This trend of greater relative similarity to T.pallidum holds for secreted proteins, with 25 best hits to pallidum and merely 3 to genitalium (out of 102 trachomatis secreted proteins). However, the majority of trachomatis prote ins are not that similar to either M.genitalium or T.pallidum.
The original concept of paralog contained the notion that functional differences would evolve after duplication -- that paralogs were not orthologs. In the absence of biochemical data, sequence analysis can't be certain that paralogous relationships wi ll be based on different functions, hence the word paralog is used in a loose sense herein and in the literature to denote similar proteins that are not thought to be orthologous. When duplicated genes with the same activity are being discussed, we will t ry to use the term isolog, leaving the term ortholog for proteins in different genomes. When paralog is used in a strict sense, different functional activities are implied. 226 of the 877 predicted proteins in C.trachomatis have similarity to one or more proteins in the organism, using a BLAST2 cutoff E value of 1e-03. These similarities are reported in the Paralog Field for each gene without any attempt to distinguish between isology and paralogy.